

The HADDOCK web server for data-driven biomolecular docking
- kübra:)

- Oct 17, 2021
- 5 min read
Biomolecular complexes are the molecular machines of the cell. To fully understand how the various units work together to fulfill their tasks, structural knowledge at an atomic level is required. Classical structural methods such as nuclear magnetic resonance (NMR) and X-ray crystallography provide this knowledge, but often encounter difficulties when it comes to complexes. Nevertheless, even in cases in which structure determination is difficult, valuable information about complexes can still be obtained from a variety of both experimental and predictive approaches. By combining this information with computational approaches such as docking, useful insights can be obtained on biomolecular interactions.
Docking
Docking is the modeling or prediction of the structure of a complex based on its known constituents. During recent years, many docking methods have been developed, including advanced methods that take into account conformational change on binding. To assess the performance of docking methods in predicting the structures of protein complexes, the Critical Assessment of Predicted Interactions (CAPRI) experiment has been set up. CAPRI has proven to act as an important catalyst in the development of biomolecular docking approaches, with the result that accurate predictions for complexes that were considered beyond the limits of docking methodology a few years ago can nowadays be obtained. Currently, the main difficulty in docking is the fact that the conformation of the starting structure (unbound structure, homology model) may be a poor approximation of the conformation in the bound form. The incorporation of structural flexibility into docking algorithms in order to deal with this problem remains an open challenge.
In docking, there are two main strategies that can be followed, namely, ab initio docking or data-driven docking. Ab initio docking only considers the coordinates of the starting structures, disregarding any experimental knowledge about the system; however, experimental information is often used to filter the docking results. In contrast, in data-driven docking, experimental or predicted information is driving the docking process directly. Among all the docking methods participating in CAPRI, only HADDOCK follows a true data-driven strategy.
HADDOCK
HADDOCK is a docking method driven by (experimental) knowledge, in the form of information about the interface region between the molecular components and/or their relative orientations. This information can be derived, e.g., from mutagenesis, mass spectrometry or a variety of NMR experiments (chemical shift perturbation (CSP), residual dipolar couplings (RDCs) or hydrogen/deuterium exchange; or, if available, classical NMR distance restraints (NOEs) can be included as well). When experimental information is sparse or absent, bioinformatic interface predictions can also be used. The 2.0 version of HADDOCK6 supports nucleic acids and small molecules; it can deal with a wide range of experimental data7 and provides improved docking protocols. HADDOCK has been applied to a variety of problems including protein–protein, protein–nucleic acid, protein–oligosaccharide and protein–small molecule complexes. Unlike many other docking programs, HADDOCK allows for conformational change of the molecules during complex formation, not only of the side chains but also of the backbone. In addition, HADDOCK directly supports the docking of NMR structures and other Protein Data Bank (PDB) structures containing multiple models.
The coordinates of more than 60 biomolecular complexes solved using HADDOCK have been deposited in the PDB11. HADDOCK has also performed very well in the CAPRI blind docking experiment (11 out of 15 complexes achieved the root mean square deviation (RMSD) <2 Å criterion for a CAPRI two-star evaluation) and is currently the most cited biomolecular docking program. The software has been licensed to over 650 laboratories around the world.
The HADDOCK docking protocol
In HADDOCK, experimental data are entered in the form of active and passive residues. These are converted by HADDOCK into Ambiguous Interaction Restraints used to drive the docking. HADDOCK automatically generates the topology of the molecules that are to be docked. The docking protocol then consists of three stages, namely, a rigid-body energy minimization, a semi-flexible refinement in torsion angle space and a final refinement in explicit solvent. After each of these stages, structures are scored and ranked, and the best structures are kept for the next stage. The HADDOCK score is a weighted sum of van der Waals, electrostatic, desolvation and restraint violation energies together with buried surface area. For every stage, the number of structures and the scoring weights can be modified by the user, as well as several parameters controlling the docking/refinement protocol such as temperature, number of time steps and force constants. HADDOCK makes use of CNS (Crystallographic and NMR system) as its structure calculation engine. In addition, HADDOCK allows the user to define the protonation states of histidines and several forms of symmetry restraints. During the torsion angle refinement, to save computation time, flexibility is only taken into account for so called semi-flexible segments and during part of the refinement. By default, these semi-flexible segments are determined automatically, by identifying the parts of the molecule that are involved in complex formation. It is possible to define manually the semi-flexible segments and also fully flexible segments that are flexible throughout the refinement. Finally, the HADDOCK program supports the simultaneous docking of up to six molecules, as any combination of proteins, nucleic acids, peptides and other biomolecules.
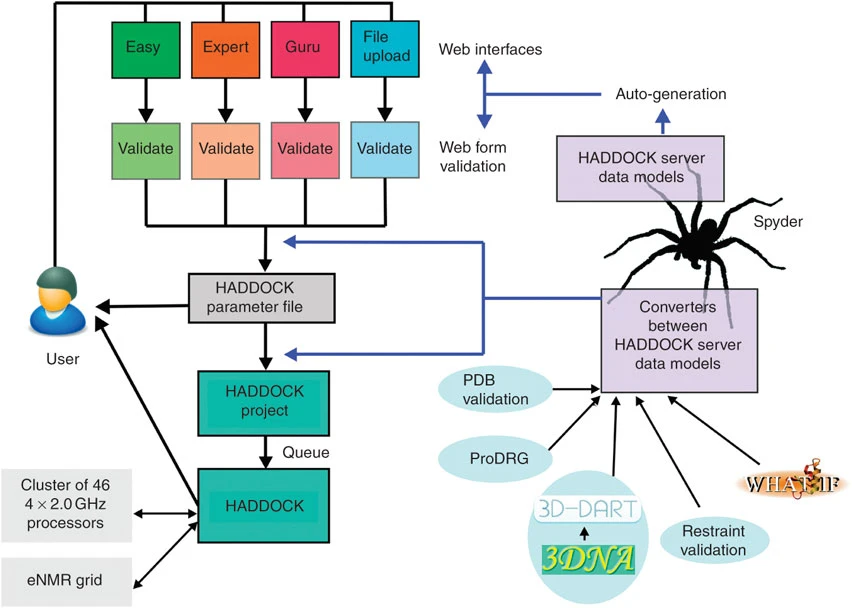
Several processing and validation routines are plugged into the HADDOCK server framework. Uploaded Protein Data Bank data are checked for correct formatting. If a structure contains unconnected multiple bodies (e.g., a dimeric receptor), restraints are automatically defined to prevent the various bodies to drift away during the docking. In case of proteins, chemical groups other than standard amino acids are detected. For a few of those groups, CNS13 parameters are already present in HADDOCK, but for all others, the PRODRG server is contacted automatically to retrieve CNS parameters. In addition, if they have not been supplied by the user, histidine protonation states are automatically defined by querying the WHATIF server. In case of nucleic acids, modules from the 3D-DART framework are used to parse and validate the supplied structures. Through its bindings to X3DNA, the framework extracts Watson–Crick base pairing from the starting structure and automatically defines restraints to preserve the helical shape of the nucleic acid during the docking. Once the data have been validated, the run is written on the server disk as a HADDOCK project directory and added to a queue. The user is kept informed of the current status of his/her run by email and a link to a unique web page is given in which the run can be monitored and the final results are presented. After the docking, the structures are clustered and the resulting clusters ranked according to the HADDOCK score. A web page containing cluster statistics is generated, from which the user can download the top four members of each clusters or preview them using Jmol. Optionally, the entire HADDOCK run can be downloaded for local manual analysis by the user.
S.J. De Vries, M. Van Dijk, A.M.J.J. Bonvin, The HADDOCK web server for data-driven biomolecular docking, Nature Protocols. 5 (2010) 883–897. doi:10.1038/nprot.2010.32.



Comments