

On Drug-Membrane Permeability of Antivirals for SARS-CoV-2
- kübra:)

- Jun 6, 2021
- 5 min read
One of the key parameters required to identify effective drugs is membrane permeability, as a compound intended for an intracellular target with poor permeability will have low efficacy. In this paper, they leverage a computational approach recently developed by their group to study the interactions between nanoparticles and mammalian membranes to study the time of the entry of a variety of drugs into the viral envelope of coronavirus as well as cellular organelles. Using a combination of all-atoms molecular dynamics simulations and statistical analysis, they consider both drug characteristics and membrane properties to determine the behavior of 79 drugs and their interactions with the viral envelope, composed of the membrane and spike protein, as well as five other membranes that correspond to various mammalian compartments (a lysosome, plasma, Golgi, mitochondrial, and endoplasmic reticulum membranes).
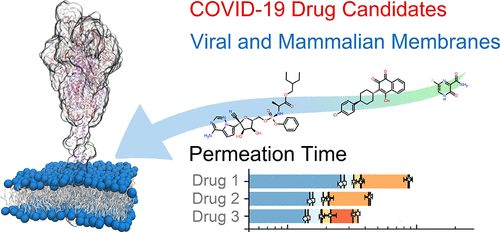
The results highlight important trends that can be exploited for drug design, from the relatively high permeability of the viral envelope and the effect of transmembrane proteins to the differences in permeability between organelles. When compared with bioavailability data present in the literature, the model results suggest a negative correlation between time of permeation and bioavailability of promising drugs. The method is general and flexible and can be employed for a variety of molecules, from small drugs to small nanoparticles, as well as a variety of biological membranes. Overall, the results indicate that this model can contribute to the identification of successful drugs as it predicts the ability of compounds to reach both intended and unintended intracellular targets.
The 2020 pandemic has brought the chasm between the rapid spread of infectious diseases and the relatively slow pace of scientific discovery to stark reality. This particular event is not the first, nor it will be the last. Since the turn of the century, three different coronavirus threats have surfaced, and emerging infectious diseases will continue to be a threat for the foreseeable future. Current methods for studying beta coronaviruses, responsible for the current pandemic, are technically and time-demanding. Viral isolation from field samples is rarely successful, and reverse genetics recovery of the recombinant virus is labor-intensive and expensive. Similarly, the design and testing of new drugs is an extremely lengthy process. To this end, drug repurposing, where existing compounds that have already been tested safe in humans are redeployed to combat another disease, has emerged as a critical strategy. As an example, Remdesivir, one of the promising COVID-19 drug candidates, is a broad-spectrum drug that was originally targeted toward a number of viruses, such as SARS-CoV and MERS-CoV, but it has also been studied to counter Ebola virus infection.
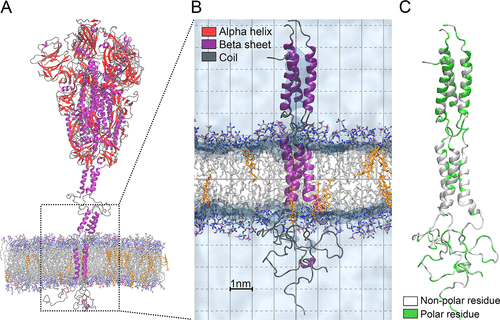
Structural diagram of the coronavirus membrane with the anchored COVID-19 spike protein. (A) Protein colored according to secondary structure (purple for α helix, red for β sheet, and gray for random coils). In the lipid bilayer, carbon atoms are shown in gray, nitrogen in red, oxygen in blue, and phosphate in tan; cholesterol is shown uniformly in orange. Hydrogen atoms are omitted for clarity. (B) The anchoring region (part of the heptad repeated, the transmembrane region, and the C-tails) of the spike protein in the membrane. (C) The anchoring region of the spike protein colored by the residue type. Nonpolar residues are colored in white, while polar residues are in green. Images generated with VMD.
Multiple characteristics define the potency of a drug, as well as its side effects. Among them, bioavailability, the fraction of a compound that reaches the intended target, is one of the hardest to estimate reliably and rapidly. Although several factors contribute to defining bioavailability, passive permeability through different biological membranes is a critical component since the inability to reach the intended target often translates into poor in vivo efficacy and may require higher dosages with higher risks of side effects.
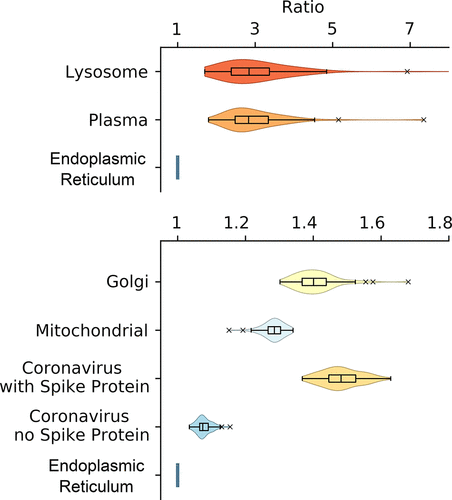
Times of entry ratios of 79 drugs in different membranes relative to the ER membrane.
The viral lifecycle encompasses several crucial steps, starting with the attachment of the virus to the cell and finishing with the release of the progeny virions from the host cell. SARS-CoV-2 targets cells through the viral structural spike protein that binds to an enzyme on the host cell membranes. Following receptor binding, the virus uses host cell receptors and endosomes to enter cells. Once inside, viral polyproteins are synthesized that encode for the replicase-transcriptase complex. The virus then synthesizes RNA and structural proteins, leading to completion of assembly and release of viral particles. These viral lifecycle steps provide potential targets for drug therapy, and most of the drugs currently in clinical trials inhibit key components of the infection lifecycle, such as viral entry into the host cell, viral replication, and viral RNA synthesis.These targets are localized to particular subcellular compartments, yet current drug design strategies are focused on general bioavailability and rarely address drug delivery to specific tissues or intracellular compartments. Therefore, knowledge of the permeability of antiviral drugs, with reference to both viral and mammalian cell compartments, is vitally important from both a pharmacokinetics and drug design standpoint. In an effort to accelerate translation of promising drug candidates, as well as to eliminate potentially ineffective or even harmful candidates, they present a science-based model to predict the permeation of drug candidates for SARS-CoV-2 into coronavirus membrane and mammalian membranes.
Over the past decades many models have been developed to estimate the permeability of drugs. The most commonly used ones are the empirical “rule of five”, QSAR (quantitative structure–activity relationship), and QSPR (quantitative structure–property relationship) methods.
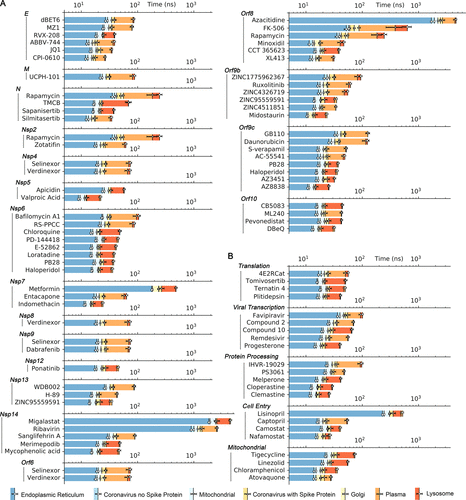
Times of permeation for drugs candidates in different organelle membranes, grouped by (A) viral baits and (B) related mammalian processes when viral baits were unknown. Error bars represent one standard deviation.
While QSAR/QSPR methods have shown their efficacy in screening the binding of antiviral drugs and targets, the predicted permeability is at best only accurate to an order of magnitude, mostly due to the incorporation of too many descriptors that are not related to the permeation process and the strong dependence on the quality of the training sets employed. On the other end of the accuracy spectrum, several methods have been proposed over the years that leverage a physical description of the passive permeation process, like the inhomogeneous solubility diffusion model and state-transition model. These models, which currently often involve biased molecular dynamics (MD) simulations, are either computationally too expensive for a rapid application or suffer from limited accuracy when drugs of various sizes and shapes or membranes with different lipid profiles are involved.
To bridge the gap between accuracy and ease of use, they have recently introduced a model to predict the rates of passive permeation of molecules and nanoparticles (up to several nanometers) into phosphatidylcholine membranes.
The low-density area (LDA) model combines five parameters, three of them that describe the permeant (i.e., size/shape, diffusivity, and hydrophobicity) and two that describe the membrane (i.e., the size and lifetime of the low-density regions on the membrane surface) to compute the average time required for the permeant to enter a membrane. The LDA model, which can combine both experimental and computational data, has shown accuracy equivalent to fully atomistic MD methods with a much smaller computational cost. Such a result was achieved by observing that the permeation process of small compounds is controlled by the formation of low-density areas on the membrane surface and by factorizing the permeation process into independent contributions of the membrane thermal and pressure fluctuations and characteristics of the molecules (size, shape, and lipophilicity).
Leveraging the LDA model, here they report on the permeation times of promising drug candidates through coronavirus membranes, with the overarching goal of providing inputs to help the design and selection of new or repurposed drugs. Additionally, since the targets of many drugs are located inside mammalian cells, they also report on the permeation kinetics of the same drugs through five mammalian membranes. Improvements). Seventy-nine drugs for SARS-CoV-2 were analyzed, comprising FDA-approved drugs, compounds in clinical trials, and preclinical compounds. The computational results are compared with data available in the literature on bioavailability and show the potential of the model to aid in the assessment of drugs’ permeability into a variety of organelles1.
On Drug-Membrane Permeability of Antivirals for SARS-CoV-2 Changjiang Liu, Paolo Elvati, and Angela Violi The Journal of Physical Chemistry Letters 2021 12 (5), 1384-1389 DOI: 10.1021/acs.jpclett.0c02397



Comments