

QSAR of dually-acting acetylcholinesterase inhibitors derived from tacrin
- kübra:)

- Jun 13, 2021
- 5 min read
Structure-activity relationships of dually-acting acetylcholinesterase inhibitors derived from tacrine on N-methyl-D-Aspartate receptors
Tacrine is a classic drug whose efficacy against neurodegenerative diseases is still shrouded in mystery. It seems that besides its inhibitory effect on cholinesterases, the clinical benefit is co-determined by NMDAR-antagonizing activity. Their previous data showed that the direct inhibitory effect of tacrine, as well as its 7-methoxy derivative (7-MEOTA), is ensured via a "foot-in-the-door" open-channel blockage, and that interestingly both tacrine and 7-MEOTA are slightly more potent at the GluN1/GluN2A receptors when compared with the GluN1/GluN2B receptors. Here, they report that in a series of 30 novel tacrine derivatives, designed for assessment of structure-activity relationship, blocking efficacy differs among different compounds and receptors using electrophysiology with HEK293 cells expressing the defined types of NMDARs. Selected compounds (4 and 5) potently inhibited both GluN1/GluN2A and GluN1/GluN2B receptors; other compounds (7 and 23) more effectively inhibited the GluN1/GluN2B receptors; or the GluN1/GluN2A receptors (21 and 28). QSAR study revealed statistically significant model for the data obtained for inhibition of GluN1/Glu2B at -60 mV expressed as IC50 values, and for relative inhibition of GluN1/Glu2A at +40 mV caused by a concentration of 100 mu M. The models can be utilized for a ligand-based virtual screening to detect potential candidates for inhibition of GluN1/Glu2A and/or GluN1/Glu2B subtypes. Using in vivo experiments in rats they observed that unlike MK-801, the tested novel compounds did not induce hyperlocomotion in open field, and also did not impair prepulse inhibition of startle response, suggesting minimal induction of psychotomimetic side effects. They conclude that tacrine derivatives are promising compounds since they are centrally available subtype-specific inhibitors of the NMDARs without detrimental behavioral side-effects. (C) 2021 The Author(s). Published by Elsevier Masson SAS.
The so-called multitarget-directed ligand (MTDL) paradigm has been widely applied in the last decade to find novel drug candidates against Alzheimer’s disease. However, this approach seems to be limited by oversimplified design of novel drug candidates combining pharmacophores with incompatible mechanisms of action, or by reason of irrelevance in the context of disease progression in time. Another limitation is associated with the fact that simple linking of the two pharmacophores usually leads to lower drug-likeness, and hence fused or merged strategies are more preferred. Since AD is currently treated by acetylcholinesterase inhibitors (AChEI) and memantine, an antagonist of N-methyl-d-aspartate receptors (NMDAR), a combination of such drugs makes sense, given the fact that impairment of both cholinergic and glutamatergic neurotransmission occurs simultaneously, i.e. in the latter stage of the disease.
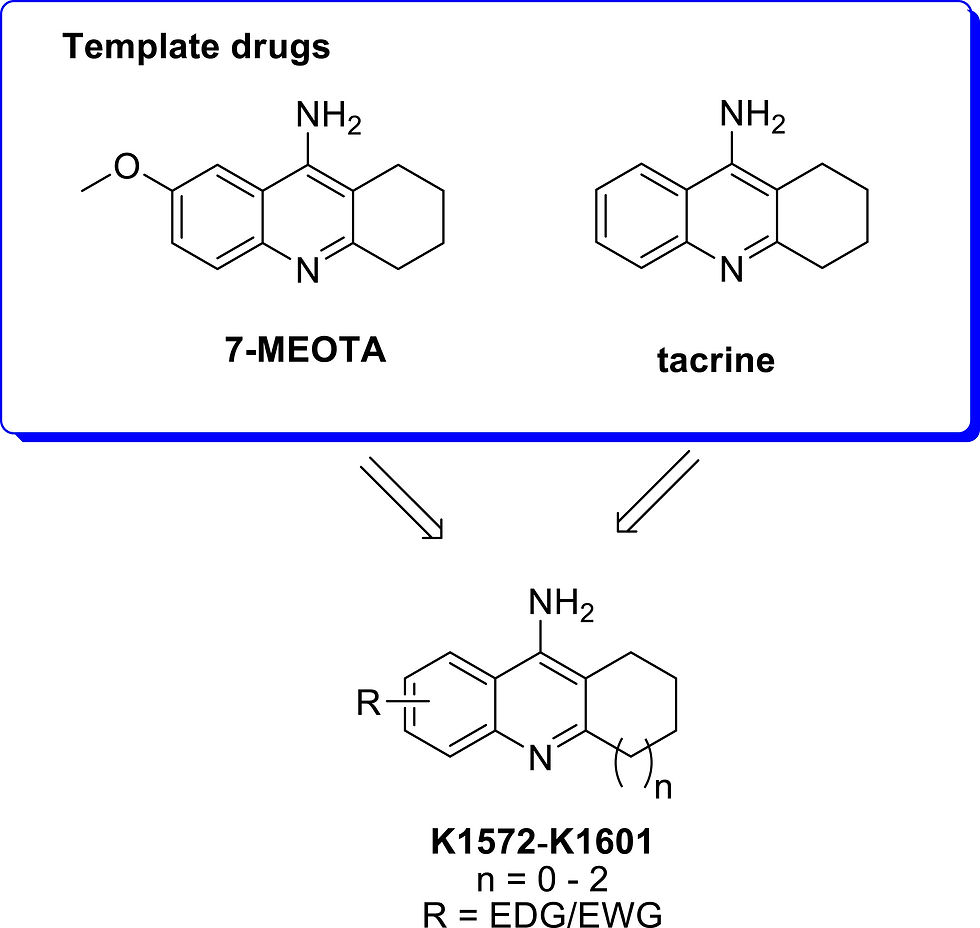
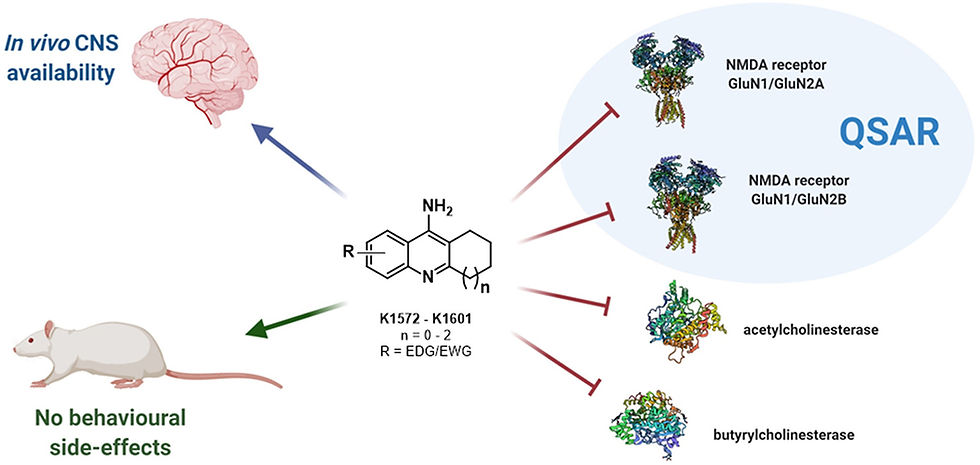
Design considerations for novel tacrine derivatives K1572–K1601 possessing dual AChE and NMDAR antagonism properties with tacrine and 7-MEOTA as template drugs.
Hence, Namzaric, a fixed dose combination of donepezil and memantine, was approved by the FDA in 2014. The dual concept applying inhibition of both AChE and NMDAR based on a linking MTDL approach was first pioneered by Simoni et al. who linked galantamine and memantine by a six-membered carbon chain to deliver memagal as a lead structure. It was followed by an in vivo study of the shorter-tethered analogue ARN14140, which demonstrated efficacy in preventing cognitive impairment and in its neuroprotective potential. In other research, tacrine-based hybrids with memantine were prepared and evaluated for their in vitro affinity towards NMDAR, and later neuroprotective efficacy was confirmed in the NMDA-induced lesion rat model for the 6-chlorotacrine – memantine hybrid. Notably, the concomitant effect of NMDAR-antagonism and AChE inhibition was established for huperzine-A, a compound approved for AD treatment in China. However, despite high expectations, its dually-acting derivatives together with bis(7)-tacrine [bis(7)-cognitin] have never reached clinical trials.
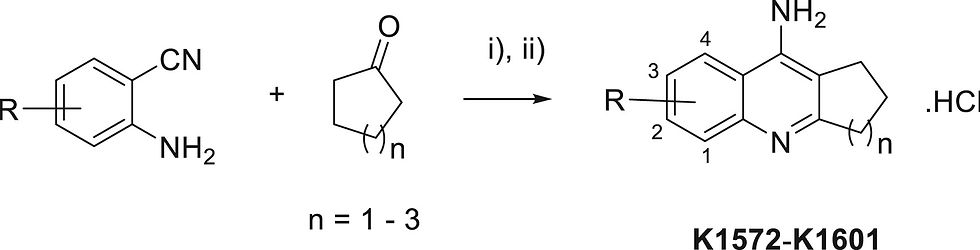
The reaction of 2-aminobenzonitrile with cyclic ketone leading to the final tacrine derivatives. ZnCl2 or AlCl3 were used as LAs in the Friedländer type condensation.
They have recently summarized that tacrine itself possesses both direct and indirect effects on glutamatergic neurons. Indirect beneficial effect may involve inhibition of Ca2+-activated potassium channels, which prevents membrane repolarization and thus leads to prolonged NMDAR activation and long-term potentiation. Interestingly, the direct effect of tacrine, as well as its 7-methoxy derivative (7-MEOTA), is implemented by inhibition of NMDAR via a “foot-in-the-door” open-channel block, with affinity in the case of 7-MEOTA comparable to that of memantine.
They also found that the IC50 values for tacrine and 7-MEOTA exhibit the following GluN2 subunit-dependent pattern: GluN1/GluN2A < GluN1/GluN2B < GluN1/GluN2C = GluN1/GluN2D. Interestingly, 7-MEOTA significantly surpassed the neuroprotective effect of both tacrine and memantine in the NMDA-induced lesion rat model, which supports a potential clinical impact.
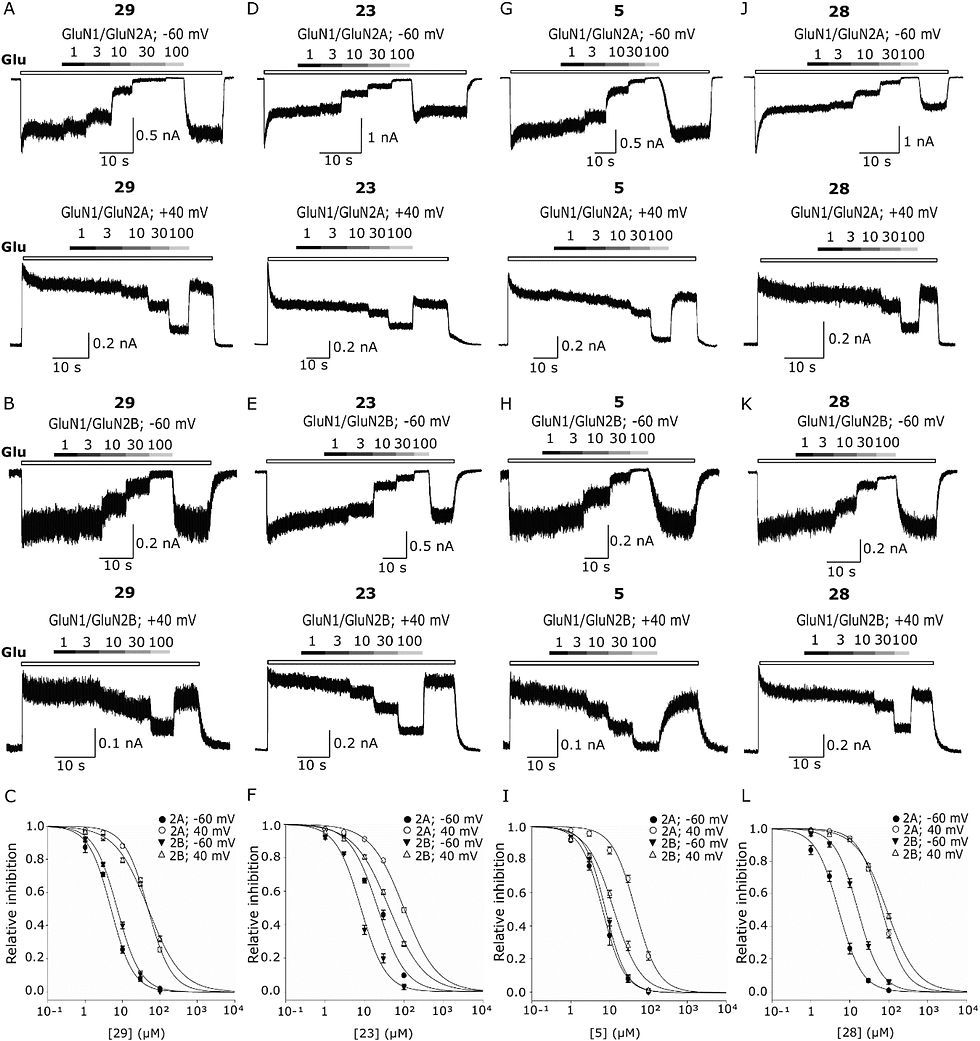
The selected most potent derivatives of tacrine. Representative responses of tacrine’s derivatives at membrane potentials of −60 mV and +40 mV from HEK293 cells transfected with GluN1/GluN2A (A,D,G,J) and GluN1/GluN2B (B,E,H,K) receptors; the identities of the derivatives are shown above each current trace with a concentration scale (1–100 μM) with co-application of 1 mM glutamate (Glu). Concentration-inhibition curves for 29 (C), 23 (F), 5 (I), 28 (L) were obtained by fitting the experimental data from GluN1/GluN2A and GluN1/GluN2B receptors at −60 and + 40 mV.
In summary, they hypothesize that the clinical efficacy of tacrine is also co-determined by the NMDAR-antagonizing activity. Thus, compounds structurally related to tacrine can be considered as true MTDLs, and tacrine’s structural simplicity ensures its drug-likeness, in contrast to MTDLs created by the linked approach. From this standpoint, different tacrine substitutions will deliver various effects on both AChE and NMDAR. In particular, effects on the latter offer a novel approach, and tuning the efficacy and selectivity to different subtypes of NMDARs represents an interesting opportunity in the field of NMDAR antagonists. It is of note that specific inhibitors of GluN1/GluN2B receptors are of interest for their suppression of the negative effects of excitotoxicity and ischemia . Moreover, it has been anticipated that low expression levels of GluN1/GluN2B receptors in the cerebellum may prevent their side effects.
The aim of this study was to develop novel tacrine derivatives with dual effect on cholinesterases and NMDAR, specifically with preference towards GluN1/GluN2A and/or GluN1/GluN2B receptors. Specifically, they synthesized a series of 30 tacrine derivatives and investigated their inhibitory potency towards human recombinant AChE (hAChE) and human plasmatic butyrylcholinesterase (hBChE), and their ability to block GluN1/GluN2A and GluN1/GluN2B receptors at negative and positive membrane potentials. These experiments were followed by analyses of quantitative structure-activity relationships (QSAR) which to the best of their knowledge was performed for the first time ever for tacrine-based compounds. In addition, to follow the potential clinical application of such dually-acting compounds, they have selected the six most promising candidates with more or less balanced activities and characterized them for their ability to cross the blood-brain barrier and for their safety in vivo, since a major concern for NMDAR ligands is their psychotomimetic side effects1.
Gorecki L, Misiachna A, Damborsky J, Dolezal R, Korabecny J, Cejkova L, et al.. Structure-activity relationships of dually-acting acetylcholinesterase inhibitors derived from tacrine on N-methyl-d-Aspartate receptors. European Journal of Medicinal Chemistry 2021;219:113434. doi:10.1016/j.ejmech.2021.113434.



Comments