

GNINA 1.0: molecular docking with deep learning
- kübra:)

- Jun 27, 2021
- 6 min read
Molecular docking computationally predicts the conformation of a small molecule when binding to a receptor. Scoring functions are a vital piece of any molecular docking pipeline as they determine the fitness of sampled poses. Here they describe and evaluate the 1.0 release of the Gnina docking software, which utilizes an ensemble of convolutional neural networks (CNNs) as a scoring function. They also explore an array of parameter values for Gnina 1.0 to optimize docking performance and computational cost. Docking performance, as evaluated by the percentage of targets where the top pose is better than 2Å root mean square deviation (Top1), is compared to AutoDock Vina scoring when utilizing explicitly defined binding pockets or whole protein docking. GNINA, utilizing a CNN scoring function to rescore the output poses, outperforms AutoDock Vina scoring on redocking and cross-docking tasks when the binding pocket is defined (Top1 increases from 58% to 73% and from 27% to 37%, respectively) and when the whole protein defines the binding pocket (Top1 increases from 31% to 38% and from 12% to 16%, respectively). The derived ensemble of CNNs generalizes to unseen proteins and ligands and produces scores that correlate well with the root mean square deviation to the known binding pose. They provide the 1.0 version of GNINA under an open source license for use as a molecular docking tool at https://github.com/gnina/gnina.
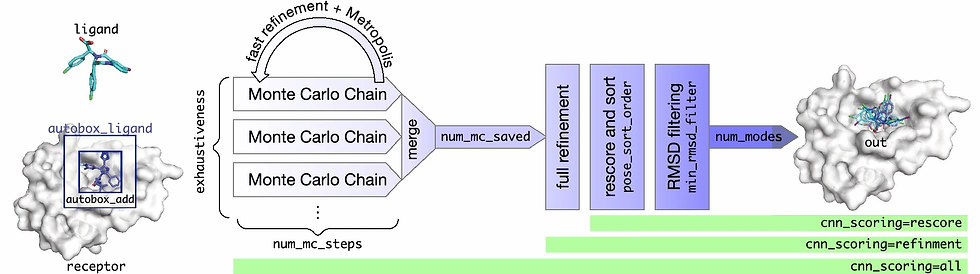
Molecular docking is a computational procedure in which the non-covalent bonding of molecules, e.g. a protein receptor and a ligand, is predicted. This prediction outputs the conformation and, usually, the binding affinity of the small molecule in its predicted minimal energy state and is used to virtually screen large libraries of compounds. Docking is composed of two main steps: sampling and scoring. Sampling refers to an extensive search of the conformational space of the molecules being docked. This conformational space is vast, due in part to both the receptor and ligand being flexible allowing for each molecule to adjust its shape due to the influence of the other. In order to constrain this large conformational space, the receptor is typically kept rigid. The other vital piece of molecular docking is the scoring function. Every sampled pose is evaluated by the scoring function for its fitness. The fitness determines the conformations that are retained from sampling and is used to rank the retained poses in the order of their likelihood of being correct. The final output of docking is a set of ranked poses of the docked molecule. Determination of the correct binding pose of a small molecule is a prerequisite for determining its binding affinity and affords the opportunity to utilize the pose for lead optimization. Correct evaluation of binding affinity is critical for downstream tasks such as virtual screening or for determining if a compound is important for more experimental analysis. Molecular docking must compute a pose and a binding affinity quickly for it to beneficial when millions of ligands are being queried in a drug discovery pipeline. Sampling the entire conformational space of a molecule is computationally demanding; therefore, they compromise on the speed and accuracy of docking to provide poses that are close to native while not requiring the full search of conformational space. This compromise requires docking software to focus on the accuracy and ranking power of the scoring function to highlight low energy conformations and reduce extraneous sampling. Scoring functions provide a mapping from the conformational space of the ligand and receptor to the set of real numbers so that poses may be ranked.

Typically, scoring functions are grouped into three categories; knowledge based, physics based, and empirical. Knowledge based scoring functions leverage the statistics from a set of structural binding data. A number of geometric properties are computed from structures of protein-ligand complexes, such as atom-atom pairwise contacts. The calculated frequencies can be used in a method such as potential of mean force (PMF), which creates a potential based on the Boltzmann distribution of the properties, to calculate the score of a pose. Knowledge based scoring functions can be biased by features present in their training sets though calculations of scores are quick at test time. They require a large database of known structures and can be difficult to interpret when trying to understand a score. Physics based scoring functions, often referred to as force fields, utilize physically derived energetics of interactions to compute scores. The final score is a summation of energy terms such as Coulombic and Van der Waals forces. Accuracy of physics based scoring functions are limited by their complexity and the assumptions they place on the fundamental forces dictating interactions between molecules, though understanding of these forces is continually increasing.
Empirical scoring functions address the limitations of physics based scoring functions by using a combination of manually selected energy terms. Rather than giving each energy term identical weighting, the weights of each term are determined via a fit to experimental data. A large proportion of docking software use empirical scoring functions, including X-Score, AutoDock Vina, and ChemScore. Unlike knowledge based scoring functions, empirical and physics based scoring functions may be easily interpreted to determine the contributing factors of a given score since each energy term can be individually queried. Fitting empirical scoring function requires a plethora of experimental structural data and prevents the combination of terms from separately trained scoring functions. The three categories of scoring functions are limited to features extracted from structural information and often assume there is a linear relationship between the features and the binding affinity. AutoDock Vina (called “Vina”) utilizes an empirical scoring function explicitly tuned to structural data. The Vina scoring function is a weighted sum of atomic interactions. Steric, hydrophobic, and hydrogen bonding interactions are calculated and are adjusted by the number of rotatable bonds to account for entropic penalties. The weights of the terms were computed via a non-linear fit to structural data. Nguyen et al. show that Vina can more accurately predict the binding pose than its predecessor, AutoDock 4. Vina demonstrates the power of modelling non-linear relationships with its increased docking performance. Therefore,they search for alternative scoring functions that are able to model non-linear relationships between inputs.

Docking using the single CNN models and the newly selected Default Ensemble for rescoring the output poses. The binding pocket is defined by the known binding ligand. TopN is the percentage of targets ranked above or at N with a RMSD less than 2 Å
Machine learning (ML) represents another growing class of scoring functions. ML algorithms learn arbitrary relationships between observations and outputs while classical scoring functions assume a specific functional form . There has been considerable progress in other biomedical fields with the utilization of ML models. However, machine learning algorithms require a large amount of data to properly generalize to unseen information. The last 20 years has seen a noteworthy increase in the quantity of available protein structures. A plethora of databases annotate structural data with experimental binding affinity data, including PDBbind and BindingDB. This information has been utilized to leverage machine learning algorithms as scoring functions. A number of traditional ML approaches have been used as scoring functions, including random forests support vector machines and gradient boosted decision trees These ML methods have been able to match or exceed existing traditional scoring functions. ML methods allow a more robust fit to training data, but are limited to features manually extracted from structural data.

Comparison between rigid and flexible docking with the default GNINA parameters: (a) ligand RMSD differences between rigid and flexible docking versus target-cognate side chain RMSDs, (b) average ligand RMSD difference for different 1 Å intervals of target-cognate side chains RMSD
Deep learning (DL) methods allow direct inference of features from inputs. They learn a representation of the inputs via layers of simple, non-linear models which transform the representation to higher abstractions to learn complex functions. DL methods have demonstrated success in a variety of fields, such as computer vision and natural language processing.
In recent years, there has been significant progress with DL methods in the drug discovery field with many models employing a convolutional framework. Convolutional neural networks (CNN) leverage convolutions to infer features directly from input tensors, usually images. CNNs have shown potential in virtual screening .and binding affinity prediction. A number of methods have been proposed to capitalize on the power of DL scoring functions. MedusaNet uses a CNN within the docking pipeline to guide the sampling of the base docking method.The base docking method, Medusa, provides a variety of ligand poses. The CNN evaluates the 3D coordinate representation of the poses to determine if a pose should be retained. Nguyen et al. describe a generative adversarial network (GAN) for pose prediction. Their network utilizes an encoder with low-dimensional mathematical representations of the protein-ligand complex and a decoder utilizing convolutional layers to generate and rank ligand poses for the D3R grand challenge. Masuda et al.use a receptor structure as the prior to their GAN to sample novel ligands appropriate to the identified binding site.
Previous work has largely evaluated deep learning protein-ligand scoring on already generated poses. In this work, they describe and comprehensively evaluate version 1.0 of the GNINA molecular docking software, a fork of SMINA and AutoDock Vina that supports CNN scoring as an integral part of the docking workflow. GNINA is evaluated here for its ability to properly score and rank binding poses for protein-ligand complexes. They describe how the default settings which balance docking accuracy and runtime were determined, including the selection of a default ensemble of CNN models. Performance of GNINA is evaluated for the redocking, cross-docking, flexible docking, and whole protein docking tasks and is found to significantly outperform SMINA/Vina in all cases.
1. Mcnutt AT, Francoeur P, Aggarwal R, Masuda T, Meli R, Ragoza M, et al.. GNINA 1.0: molecular docking with deep learning. Journal of Cheminformatics. Journal of Cheminformatics; 2021;13(1).



Comments