

Generative Deep Learning for Targeted Compound Design
- kübra:)

- Dec 12, 2021
- 6 min read
In the past few years, de novo molecular design has increasingly been using generative models from the emergent field of Deep Learning, proposing novel compounds that are likely to possess desired properties or activities. De novo molecular design finds applications in different fields ranging from drug discovery and materials sciences to biotechnology. A panoply of deep generative models, including architectures as Recurrent Neural Networks, Autoencoders, and Generative Adversarial Networks, can be trained on existing data sets and provide for the generation of novel compounds. Typically, the new compounds follow the same underlying statistical distributions of properties exhibited on the training data set Additionally, different optimization strategies, including transfer learning, Bayesian optimization, reinforcement learning, and conditional generation, can direct the generation process toward desired aims, regarding their biological activities, synthesis processes or chemical features. Given the recent emergence of these technologies and their relevance, this work presents a systematic and critical review on deep generative models and related optimization methods for targeted compound design, and their applications.

De novo molecular design aims to create new chemical entities with desired properties and/or activities. These properties may be easily quantifiable, such as molecular weight, or somewhat more abstract, as is the case of toxicity. This is an inherently difficult task owing to the immense search space of around 1033–1080 feasible molecules from which only a small fraction typically have the desired traits. As such, de novo molecular design was, for many years, and mostly remains a process of almost exclusive trial and error, with human expert knowledge and intuition about chemistry playing a major role.
Meanwhile, the high costs associated with developing new molecules, reaching $2.8 billion dollars for a single compound, have also led to the implementation of computational tools capable of assisting the process. These have proven valuable and have found wide usage in practical applications. A forthright approach consists in enumerating all possible molecules that conform to valency rules and do not include chemically unstable functional groups. A notable example is the Chemical Space project, where this technique was employed to generate 166 billion molecules.Another technique, reaction-based de novo design, uses a set of known chemical reactions to combine various readily available building blocks into new molecules. This process can be guided by a similarity criterion to a known molecule of interest, giving rise to a large number of new similar molecules while ensuring their synthetic plausibility.
Evolutionary Algorithms (EAs) have also been successfully applied to de novo molecular design. As a recent example, AutoGrow4 uses an EA to create new predicted ligands. At each iteration, new molecules are created using a mutation operator, that performs an in silico chemical reaction, or a crossover operator that merges two compounds into a new one by randomly combining their decorating moieties. Grammatical Evolution on string representations and evolving molecular graphs provide alternative approaches that enable EAs to generate novel compounds targeting desired properties.

Acetaminophen (center) under various molecular representations. Top-left: Sequence based representations. Prior to being fed to the models, these sequences are also usually one-hot encoded. Top-right: Graph-based representations. While connection matrices are a suitable input for standard architectures, graphs can also be directly handled using graph neural networks. Bottom: Three dimensional representations, images from PubChem. Graphs may be enhanced by including 3D information as node attributes, such as internal distances and angles, or based on a coordinate system such as Cartesian space. Molecular surfaces can be voxelized into a 3D grid for easier processing.
Although useful, these methods still leave room for improvement. For instance, enumeration often leads to molecules that are too difficult to synthesize, and reaction-based design is fundamentally restricted in its ability to explore the chemical space, both important aspects of molecular design. EAs, while computationally efficient and capable of performing on par with other recent approaches, rely on expertly encoded operations, possibly limiting the search space and not leveraging the large amounts of data currently available.More recently, advances in Deep Learning (DL) sparked a surge of novel approaches to these problems. Over the past decade, DL has proven successful in multiple fields, such as computer vision, speech recognition, and translation, pushing the state-of-the-art forward and surpassing other Machine Learning approaches. DL refers to the use of artificial neural networks with multiple hidden layers. A common intuition is that successive layers can learn higher-level abstractions of the inputs.

Top-left: Three layer Recurrent Neural Network (RNN) both rolled and unrolled. In each layer, the output of a step, besides flowing to the next layer, also flows to the next step of the layer itself. These recurrent connections are depicted in the unfolded view of the network as vertical arrows. Top-right: Variational Autoencoder (VAE) where the input is encoded to the parameters of a statistical distribution, namely, the means (μ) and standard deviation (σ). In practice, these correspond to two vectors which, on the sampling step, are interpreted as a set of means and standard deviations. Bottom-left: Generative Adversarial Network (GAN) composed by a generator and a discriminator. Training seeks not a minimum but a useful equilibrium between the generator and the discriminator. Bottom-right: Adversarial Autoencoder (AAE) where the attached discriminator must discern between encoded points and samples drawn from a prior statistical distribution.
Within DL, generative modeling aims at capturing an underlying data generation process, some unknown probabilistic distribution from which a data set was sampled, and has been successfully used for creative tasks, such as writing, composing music, and painting. It usually deals with unlabeled data and attempts to create a model capable of generating new observations that closely resemble those from the training data and not simply producing copies.

Left: General procedure for the generation of 3D shapes as proposed by Skalic et al.The convolutional decoder of a VAE is used to produce a 3D molecular shape which is converted to SMILES by a captioning network. Right: General process for generating molecules as 3D point sets, proposed by Gebauer et al. It is conceptually similar to the sequential graph generation, operating on point sets with an internal coordinate system.
Improving on earlier approaches, which employed more traditional machine learning methods such as Gaussian Mixture Models, deep generative models have recently found use in the generation of novel molecular entities. Starting around 2017, with works like that of Ǵomez-Bombarelli et al., Yuan et al. and Segler et al., a large number of novel approaches have been put forward employing various neural network architectures and molecular representations. Alongside these works, several reviews have also sought to condense the plethora of different approaches, shedding light into and discussing the different architectures, generation of various molecular representations and use of comparative metrics.
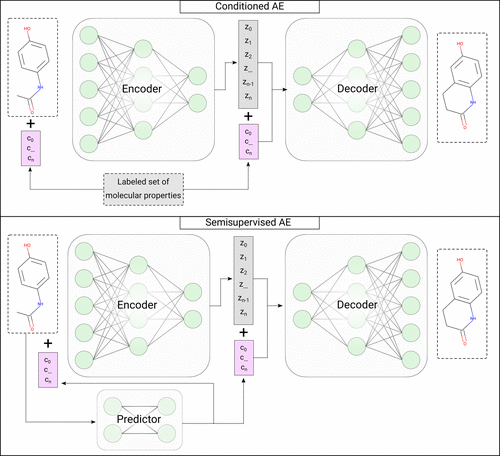
Top: In conditioned generation, the desired properties are introduced as explicit inputs to the model. These properties are precomputed for each compound of the training set and used during training to induce a correlation between the two. This correlation is then leveraged during the generation process to target specific property values. Bottom: In the semisupervised case of conditioned generation, only part of the training set has the desired properties available. To overcome this, a predictor network is trained on the labeled instances and used to predict the properties of unlabeled ones.
Due to the vastness of chemical space and the costs associated with testing possible compounds, a rational exploration with regard to the desired properties is preferable. As such, a number of distinct approaches have been developed for directing and controlling the generating process of molecules toward compounds with defined chemical properties or desired activities.
In this arena, the evolution has been very fast, with novel methods appearing at an impressive rate. A systematic review of the main advances of DL in the generation of focused molecules seems particularly relevant for practitioners interested in understanding the main features of each method, their main advantages, and their limitations. Several reviews covering the recent explosion of interest in generating molecules leveraging DL have been published. These, however, have mainly focused on the various architectures and molecular representations while only briefly touching on the various methods for the targeted generation of compounds.
Particularly, Schwalbe-Koda and Ǵomez-Bombarelli mention some of these methods, but they primarily focused on the several molecular generation schemes. Likewise, several other reviews also make note of a couple of these methods while not delving into further discussion. Closer to our work, Sanchez-Lengeling and Aspuru-Guzik directly discussed the inverse molecular design problem, touching on some methods for controlling the properties of generated molecules.
Notwithstanding previous efforts on reviewing this field, they feel that a more rigorous approach to this subject, containing a more systematic coverage of the methods, can be important for researchers working on these topics. To that end, here they aim to provide a comprehensive review of DL methods for the targeted generation of novel compounds. As such, after an introduction to molecular representations, they present the most common deep generative models and the underlying neural network architectures. They, then, focus on the different optimization approaches that allow to focus the search on molecules with desired properties or activities, closing with a review of the main practical applications.
Generative Deep Learning for Targeted Compound Design Tiago Sousa, João Correia, Vítor Pereira, and Miguel Rocha Journal of Chemical Information and Modeling 2021 61 (11), 5343-5361 DOI: 10.1021/acs.jcim.0c01496



Comments